
Badania nad mikrobiomem jelit pacjentów z COVID-19, opublikowane w Frontiers in Microbiology przez zespół z Małopolskiego Centrum Biotechnologii, odkrywają, jak mikrobiom wpływa na przebieg choroby u hospitalizowanych pacjentów.
Wpływ COVID-19 na przewód pokarmowy
Pandemia COVID-19 wywołana przez SARS-CoV-2 spowodowała szeroki wachlarz objawów klinicznych, z których najczęstsze są objawy oddechowe. Jednakże pojawiające się dowody sugerują, że przewód pokarmowy jest również atakowany przez wirusa. Enzym konwertujący angiotensynę 2 (ACE2), kluczowy receptor dla wirusa SARS-CoV-2, występuje obficie w jelicie krętym i okrężnicy. Wirus SARS-CoV-2 wykrywano w tkankach przewodu pokarmowego oraz w próbkach kału, nawet w przypadkach ujemnych wyników obecności SARS-Cov-2 w drogach oddechowych. Objawy związane z przewodem pokarmowym wiązały się z zwiększonym ryzykiem hospitalizacji na oddziale intensywnej terapii oraz śmiertelnością.
Rola mikrobiomu jelitowego
Mikrobiom jelitowy, złożony ekosystem około 40 miliardów bakterii, odgrywa kluczową rolę w immunologicznych i metabolicznych ścieżkach. Obserwowano dysbiozę mikrobioty jelitowej, charakteryzującą się utratą korzystnych mikroorganizmów i zmniejszoną różnorodnością mikroorganizmów, u pacjentów z COVID-19, potencjalnie przyczyniającą się do nasilenia choroby.
Badanie mikrobiomu jelitowego w COVID-19
W publikacji ukazanej w Frontiers in Microbiology naukowcy przeanalizowali mikrobiom jelit 204 pacjentów hospitalizowanych z powodu ciężkiego przebiegu COVID-19. Celem badania było prześledzenie zmian składu mikroorganizmów w trakcie hospitalizacji oraz powiązanie tych zmian z procedurami klinicznymi (podawanie antybiotyków, przyjęcie na OIOM, wynik hospitalizacji: przeżycie bądź śmierć). Oceniono potencjał predykcyjny mikrobiomu jelitowego dla rokowania COVID-19. Wykazano zróżnicowany wpływ parametrów klinicznych, danych podstawowych pacjenta (płeć, wiek, BMI) i mikrobiomu na precyzję prognozowania wyniku hospitalizacji (skierowanie na OIOM, przeżycie/śmierć). Pokazano, że dane mikrobiomowe mają większą wiarygodność w prognozowaniu wyników pacjentów w porównaniu z danymi klinicznymi lub danymi podstawowymi.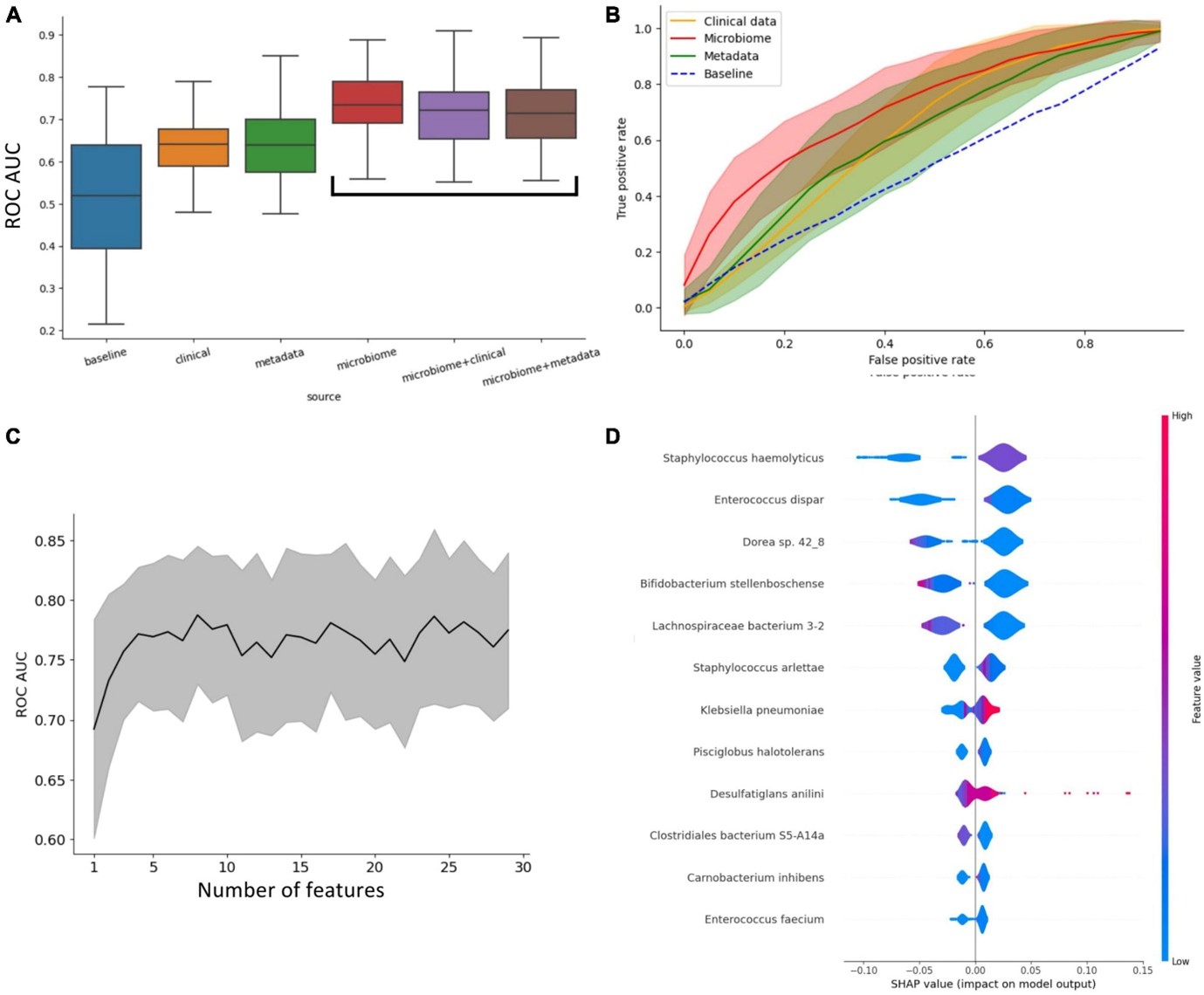
Rycina 1. Wpływ parametrów klinicznych, danych podstawowych pacjenta (płeć, wiek, BMI) i mikrobiomu na precyzję prognozowania wyniku hospitalizacji (skierowanie na OIOM vs. non-OIOM). (A) Wpływ różnych typów danych na zdolność przewidywania wyniku hospitalizacji. Dane mikrobiomowe znacząco poprawiają wyniki klasyfikatorów. (B) Krzywa ROC klasyfikatorów pogrupowana według typu danych. (C) Zwiększenie różnorodności mikrobiologicznej nie poprawia wyników ROC-AUC poza 7 istotnymi gatunkami. (D) Wartości Shapleya dla istotnych gatunków.
Porównanie metod sekwencjonowania mikrobiomu
Ostatnie badania nad mikrobiomem jelit pacjentów z COVID-19 korzystały z dwóch głównych metod sekwencjonowania: sekwencjonowania genów 16S rRNA oraz głębokiego sekwencjonowania metagenomicznego typu shotgun. Pierwsza z tych metod jest bardziej ekonomiczna i nadaje się do dużych zbiorów próbek, ale nie zapewnia tak precyzyjnej informacji o rodzajach bakterii jak głębsze sekwencjonowanie shotgun. To drugie jest bardziej dokładne, ale kosztowne, co ogranicza jego powszechne stosowanie w badaniach klinicznych. Okazuje się, że płytkie sekwencjonowanie shotgun, które jest tańsze od głębokiego, zapewnia równie dokładne wyniki w kluczowych aspektach badania mikrobiomu, co czyni je obiecującym narzędziem do zastosowań w praktyce klinicznej, szczególnie w kontekście zarządzania COVID-19.
Płytkie sekwencjonowanie shotgun jako alternatywa
Zespół dr Tomasza Kościółka wykazał, że płytkie sekwencjonowanie shotgun stanowi realną i opłacalną alternatywę diagnostyczną dla głębokiego sekwencjonowania w środowiskach klinicznych. Obserwowano wysoki stopień pokrycia gatunków zidentyfikowanych w płytkim i głębokim sekwencjonowaniu. Głębsze sekwencjonowanie ujawniło znacznie więcej gatunków, jednak wszystkie oprócz pięciu gatunków zidentyfikowanych w płytkim sekwencjonowaniu zostały także wykryte w głębokim sekwencjonowaniu. Dodatkowo pokazaliśmy, że chociaż obfitość gatunków nie była idealnie dopasowana między płytkim a głębokim sekwencjonowaniem, hierarchia obfitości gatunków, nawet w przypadku niskich obfitości poniżej 1%, była dobrze zachowana.
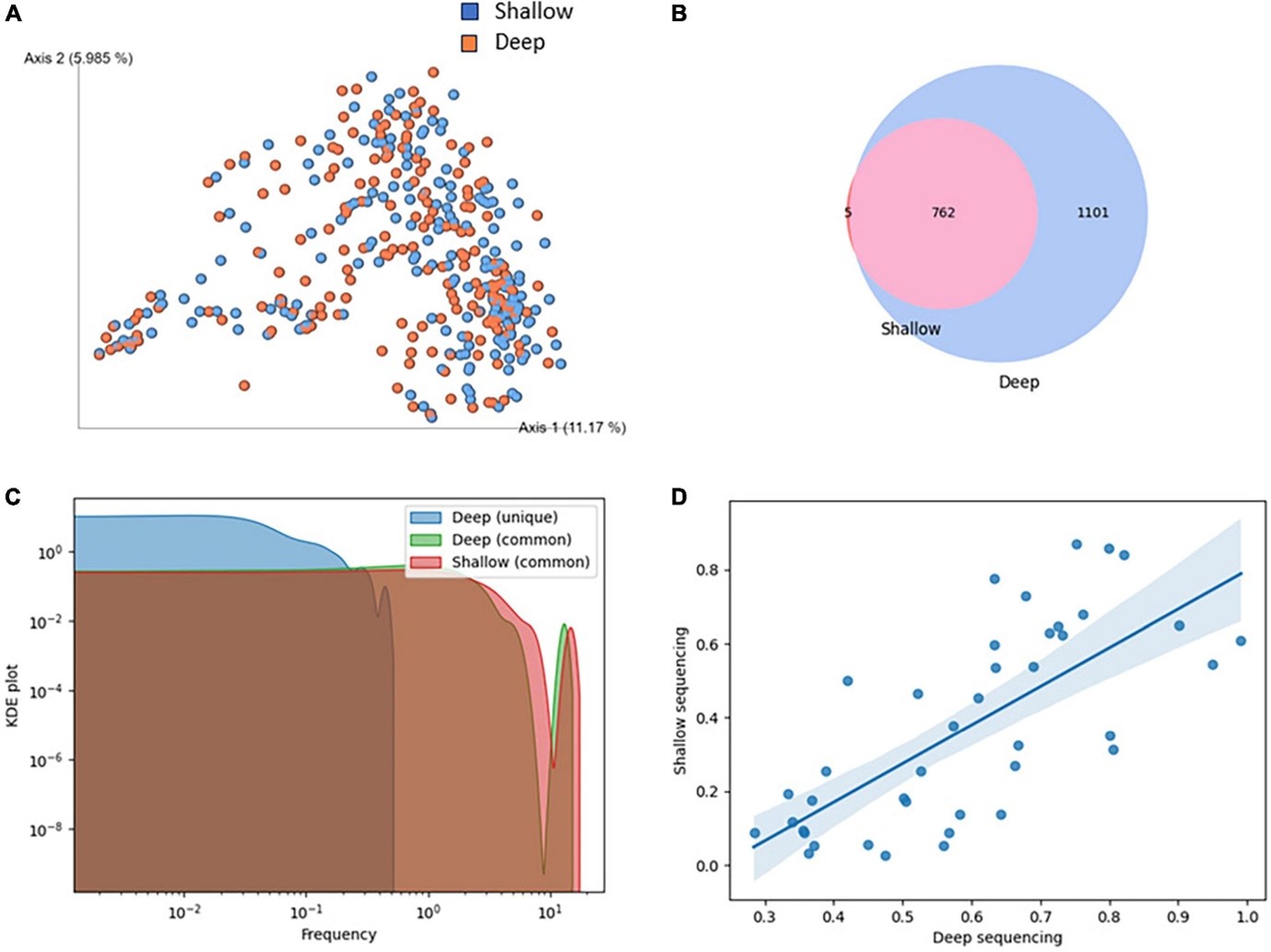
Rycina 2. Porównanie metod płytkowego i głębokiego sekwencjonowania shotgun. (A) Klastrowanie próbek na podstawie różnorodności beta Bray-Curtis. Niebieski: głębokie sekwencjonowanie, czerwony: płytkowe sekwencjonowanie. (B) Przecięcie gatunków zidentyfikowanych w płytkowym i głębokim sekwencjonowaniu. (C) Wykres KDE obfitości gatunków zidentyfikowanych wyłącznie lub wspólnie w płytkowym i głębokim sekwencjonowaniu.
Badanie prowadził zespół badaczy MCB UJ pod przewodnictwem dr Tomasza Kościółka przy wsparciu dr hab. inż. Pawła Łabaja, we współpracy z firmą Sanprobi sp. z o. o., Pomorskim Uniwersytetem Medycznym w Szczecinie oraz Państwowym Instytutem Medycznym MSWiA w Warszawie.
Unikalność danych i ich obfitość daje nadzieję na dalsze odkrycia, zwłaszcza przy poszerzonej analizie danych z głębokiego sekwencjonowania. Analiza ta jest obecnie przeprowadzana przez grupę Genomiki Strukturalnej i Funkcjonalnej dr Tomasz Kościółka (obecnie: Sano – Centrum Zindywidualizowanej Medycyny Obliczeniowej) przy współudziale badaczy MCB UJ oraz pozostałych konsorcjantów.