Naukowcy z Małopolskiego Centrum Biotechnologii Uniwersytetu Jagiellońskiego wraz z kolegami z Australii, Turcji i Kanady odkryli powiązanie między defektami w kompleksie modyfikującym cząsteczki tRNA, a zaburzeniami neurorozwojowymi (NDD), charakteryzującymi się niezdolnością do osiągnięcia poznawczych i motorycznych kamieni milowych rozwoju. Badacze wykazali, jak mutacje genetyczne u pacjentów wpływają na aktywność Elongatora i prowadzą do poważnych objawów klinicznych.
Zapewnienie odpowiedniej szybkości syntezy białek jest kluczowe dla integralności proteomu i rozwoju neuronów. Kompleks Elongatora modyfikuje cząsteczki tRNA w pozycji „chwiejnej” zasady (U34), aby zapewnić jednoznaczne rozpoznawanie kodonów podczas translacji białka. Badanie to dostarcza pierwszych dowodów klinicznych na to, że mutacje zmiany sensu w podkompleksie pomocniczym Elongatora ELP456 powodują zaburzenia neurorozwojowe. Analiza całego genomu pozwoliła na identyfikację patogennych wariantów ELP4 i ELP6 u pacjentów z ciężką postacią kliniczną NDD. Dalsze modelowanie mutacji pochodzących od pacjentów u myszy doprowadziło do odtworzenia złożonego fenotypu neurorozwojowego i ujawniło specyficzne dla neuronów konsekwencje mutacji.
„Wykazaliśmy, że substytucje znalezione u pacjentów, w podkompleksie pomocniczym ELP456, wpływają na inne typy neuronów niż znane wcześniej mutacje w rdzeniu katalitycznym kompleksu” – wyjaśnia dr hab. Sebastian Glatt, lider Grupy Badawczej Maxa Plancka, która prowadziła badania doświadczalne w Krakowie. Odkrycie stanowi nową koncepcję w tej dziedzinie, zgodnie z którą zubożenie określonych modyfikacji cząsteczek tRNA w komórkach pacjenta może indukować określone zmiany w proteomie komórkowym.
Naukowcy starali się także zrozumieć molekularne konsekwencje wyników uzyskanych z komórek pacjentów. Używając nowoczesnej mikroskopii krioelektronowej do wizualizacji cząstek makromolekularnych, badacze opracowali modele wysokiej rozdzielczości ludzkich i mysich podkompleksów ELP456 oraz zlokalizowali podstawienia aminokwasów pochodzące od pacjentów.
„Co ciekawe, nasze odkrycia podkreślają, że integralność kompleksu Elongatora in vitro i in vivo nie była zaburzona przez mutacje w genach ELP4/6, natomiast jego powinowactwo wiązania z określonymi cząsteczkami tRNA oraz jego aktywność biochemiczna były silnie zmniejszone” – mówi dr Monika Gaik, adiunkt i pierwsza współautorka artykułu.
Przedstawiona praca łączy dane kliniczne, modele zwierzęce z mutagenezą obejmującą całe ciało i badaniami biochemicznymi, w celu zrozumienia fundamentalnych podstaw roli kompleksu Elongatora w rozwoju neuronów i struktur mózgowych.
+930px.jpg/3fe3549c-bd48-4f4d-8ae1-7c62ba47a02e?t=1659340176011)
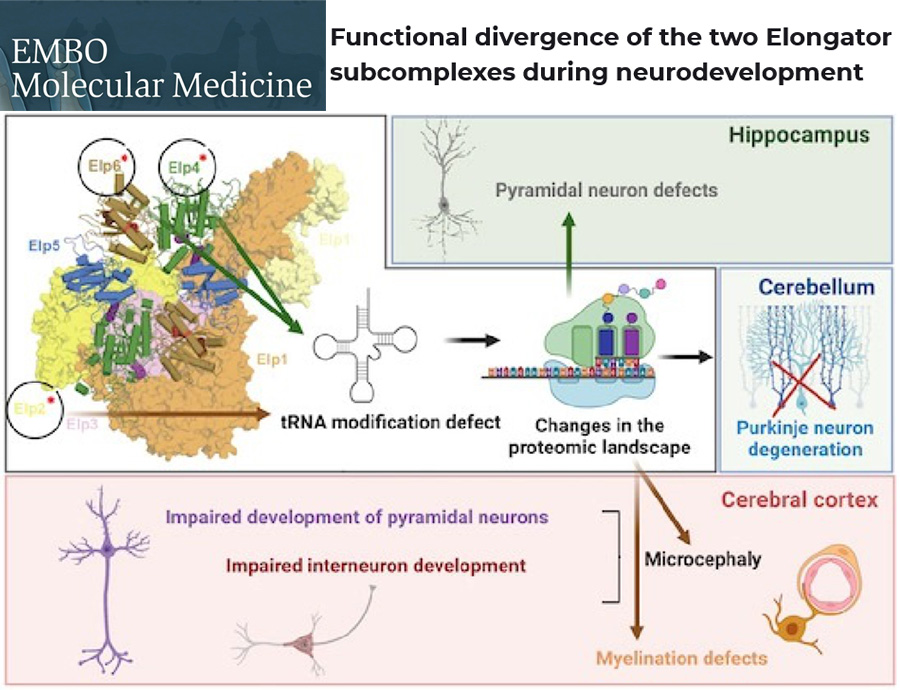
Rycina: Kompleks Elongator odgrywa kluczową rolę w rozwoju neuronów. Źródło: EMBO Molecular Medicine - https://doi.org/10.15252/emmm.202115608.
Realizacja badań w zakresie rozszerzenia niedawno powstałej koncepcji „wyspecjalizowanych rybosomów” poprzez dodanie unikalnych modyfikacji RNA w kontekście diagnozowania i leczenia rzadkich chorób neurodegeneracyjnych oraz rozwoju neuronów jest możliwa dzięki wsparciu Fundacji na Rzecz Nauki Polskiej i Komisji Europejskiej, fundatora prestiżowego ERC Consolidator Grant, który otrzymał dr hab. Sebastian Glatt.
https://www.fnp.org.pl/laureaci-fnp-zdobywcami-grantow-erc/

Projekt finansowany przez Europejską Radę ds. Badań Naukowych (ERC) w ramach programu Unii Europejskiej "Horyzont 2020" w zakresie badań naukowych i innowacji (umowa o grant nr 101001394).
Artykuł został opublikowany w EMBO Molecular Medicine [EMBO Mol Med (2022)e15608] i jest dostępny tutaj:
Functional divergence of the two Elongator subcomplexes during neurodevelopment